by Preston Parana
Nominated by Angeliz Soto Acevedo for CHEM 353: Introduction to Biochemical Research Techniques and Scientific Writing
Instructor Introduction
Throughout the semester, students worked on developing a paper on a protein of their interest. As Preston presented his work, Succinate Dehydrogenase: The Stylistic Interconnection of Aerobic Metabolism, a collective appreciation ensued. Preston’s paper stood out as an informative, clearly written, and engaging piece of work that elegantly describes complex biochemical features and processes as well as applications and relevant clinical aspects related to the protein succinate dehydrogenase. Moreover, Preston’s paper conveyed these topics with remarkable structural flow and content robustness. Without a doubt, Preston’s paper is an outstandingly well-written piece of work that is highly meritorious for the Sweetland Upper-Level Writing Prize.
— Angeliz Soto Acevedo
Succinate Dehydrogenase: The Stylistic Interconnection of Aerobic Metabolism
Abstract
Throughout the past century, biochemists have sought to elucidate how aerobic metabolic pathways in the mitochondria of eukaryotes stylistically interconnect. This review highlights the only enzyme that exists in both the citric acid cycle and the electron transport chain: succinate dehydrogenase (SDH). As an oxidoreductase, SDH catalyzes the oxidation of succinate to fumarate and the reduction of cofactor FAD to energy carrier FADH2. X-ray crystallography has shown that SDH is a heterotetrameric, transmembrane protein complex bound to the inner mitochondrial membrane, where SDHA and SDHB make up the catalytic domain while SDHC and SDHD form the transmembrane domain. Although the presence of bound cofactors and prosthetic groups ensures proper SDH function, many compounds reversibly and competitively bind SDH to block electron transport. These inhibitors, called SDHIs, have been shown to not only treat fungal disease in various crops, but also to disrupt ecological population stability. While SDH inhibition is not well studied in humans, SDH disruption via deleterious mutation may result in the onset of diseases like paraganglioma, Leigh syndrome and clear cell renal cell carcinoma. This indicates that there is potential for SDH as the target point for cancer therapy, such that its inhibition would slow uncontrolled tumorigenesis.
Introduction
Essential for the survival of most eukaryotic organisms, aerobic metabolism is the process by which the body produces energy, in the form of ATP, using oxygen. The mitochondria catalyzes several aerobic metabolic processes, including the citric acid cycle, that converts acetyl-CoA to oxaloacetate and various energy carriers, and the electron transport chain (ETC), that utilizes these energy carriers to create ATP for cellular use. Succinate dehydrogenase (SDH) is a unique enzyme such that it can participate in both the ETC and the citric acid cycle.1 Specifically, SDH is a 129.2 kDa oxidoreductase bound to the inner mitochondrial membrane of eukaryotes, and it catalyzes the sixth step of the citric acid cycle: the oxidation of succinate to fumarate.1 This reaction simultaneously reduces FAD to FADH2, an energy carrier that is ultimately used to reduce coenzyme Q to ubiquinol.2 Once ubiquinol is created, it travels to complex III in the ETC to participate in the Q cycle and develop a proton gradient across the membrane.3 Without SDH, the citric acid cycle would not be able to continue, thereby halting the production of energy carriers such as NADH and FADH2 that are essential for continued function of the ETC. Although the ETC would not be disrupted entirely, ATP production in SDH deficient cells would decrease significantly compared to those that contain functional SDH, effectively depleting the energy available for other cellular processes.4 This suggests that eukaryotes have developed stylistic and interconnected aerobic metabolic pathways through unique enzymes like succinate dehydrogenase.
Although the first assessment of SDH structure and function surged from biochemists in the 1950s, the rapid development of research dedicated to intermediary metabolism has allowed for many studies to further characterize the role of SDH in humans.2 This review will first focus on the heterotetrameric structure of SDH by considering the role of its four different subunits and highlighting significant ligand binding sites, cofactors, prosthetic groups, and intermolecular forces that contribute to its function. Furthermore, this review will explore the structure and binding of SDH’s various inhibitors, including atpenin A5, in order to elucidate their agricultural significance and toxicity to human physiological systems. Lastly, SDH deficiency due to subunit-specific mutations has been shown to not only lead to growth inhibition but also rare neuroendocrine and neurometabolic human diseases, such as paraganglioma and encephalopathy.2 Thus, this review will seek to characterize common mutations in SDH associated with disease, explain how mutation of functional SDH affects the citric acid cycle and ETC, and recognize the public health implications of SDH mutation.
Succinate Dehydrogenase Heterotetrameric Structure & Function
Succinate dehydrogenase is a heterotetrameric, transmembrane protein complex that weighs 129.2 kDa (encoded by the Gallus gallus avian nuclear genome) and is composed of four structurally different subunits: SDHA, SDHB, SDHC, and SDHD.1 The crystal structure of SDH, discovered by X-ray crystallography and diffraction experiments, shows that the SDHA and SDHB subunits form the catalytic domain, are both hydrophilic, and protrude into the mitochondrial matrix.5 SDHA is 621 amino acids long and weighs 70.9 kDa.6 It is a flavoprotein due to the fact that it contains a covalently bound FAD prosthetic group that becomes converted to FADH2 when succinate interacts with its binding site and becomes oxidized to fumarate in the citric acid cycle, as seen in Figure 1.5 Specifically, succinate is stabilized at its binding site by the polar side chains of various SDHA residues through hydrogen bonds.7 Crystal structure also suggests that FAD is bound by a linkage to residue H56 at the succinate binding site and is further coordinated by hydrogen bonds from the side chains of nearby polar amino acids.6 One study shows that this linkage is conserved across eukaryotic species, plays a role in electron transfer to SDHB and iron-sulfur clusters, and allows for discrimination between apo- and holo-SDHA protein.8 SDHB is an iron-sulfur protein with three iron-sulfur prosthetic groups for electron transfer to ubiquinone molecules, as shown in Figure 1.5 It weighs 30.1 kDa and is composed of 252 amino acids.6 Covalent linkage to SDHB occurs when local cysteine residues become oxidized to form disulfide bonds with the sulfur atoms within the Fe-S clusters.6 As a result, electrons are released and can be transferred for other redox reactions, like reduction of ubiquinone at its binding site in SDHC/SDHD.1 Thus, SDHB tends to interact with the transmembrane subunits of SDHC and SDHD (Figure 1).9
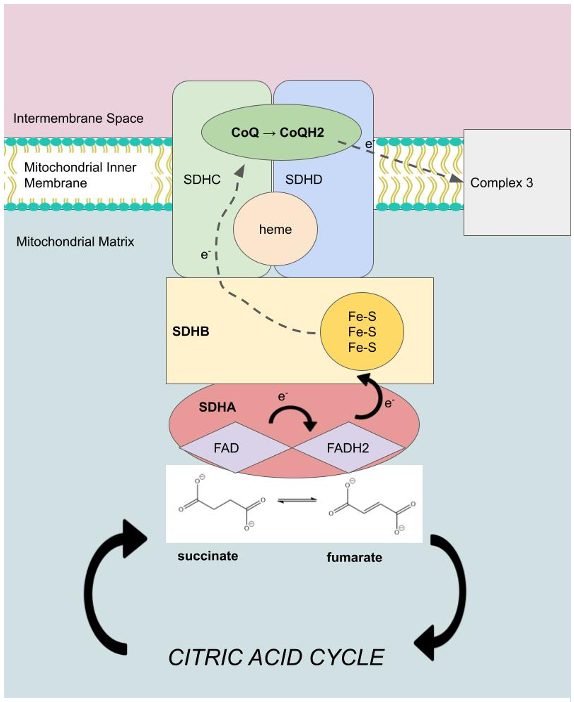
SDHC/SDHD are hydrophobic, integral proteins that anchor the SDH protein complex to the inner mitochondrial membrane.5 The SDHC gene produces an 16.7 kDa protein composed of 141 amino acids, while the SDHD gene produces a 11.5 kDa protein composed of 103 amino acids.5 These subunits contain six transmembrane helices, a ubiquinone binding site for the conversion of ubiquinone to ubiquinol, and one heme group.5 Heme is a prosthetic group in SDHC/SDHD that has been shown to not only bind oxygen but also enhance electron transport and allow for SDH protein assembly.10 This demonstrates that the succinate binding site in SDHA and the ubiquinone binding site in SDHC/SDHD ultimately become connected by a chain of different redox centers across a distance of over 40 Å in the enzyme, including FAD, the Fe-S clusters, and heme.6 Because all edge-to-edge distances are less than the suggested 14 Å limit for physiological electron transfer, succinate dehydrogenase’s structure allows for proper function of the ETC.11 Once ubiquinone is reduced, its electrons are transferred to Complex III to further establish the proton gradient (Figure 1).12
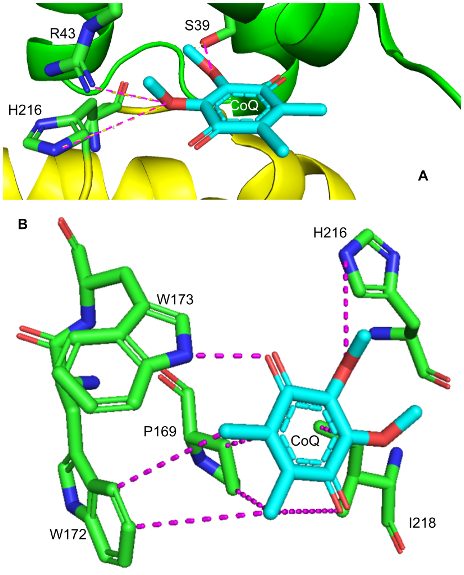
As shown in Figure 2, the ubiquinone binding site is found in a gap that is surrounded by SDHB, SDHC and SDHD. Ubiquinone, also called coenzyme Q (CoQ) is a non-aromatic, cyclic ligand that has both a polar face, characterized by its ether and ketone functional groups, as well as a nonpolar face, with its cycloalkyl groups. These faces make favorable interactions with the side chains of several amino acids from the surrounding subunits. For example, the polar face of CoQ interacts with polar hydrophilic residues of SDHB (H216) and SDHC (S39, R43).13 The short distance between hydrogen bond donors and acceptors allows for stabilizing interactions between these polar molecules and contributes to efficient binding of the polar face of CoQ.13 On the other hand, the nonpolar face of CoQ favorably interacts with nonpolar hydrophobic residues of SDHB (P169, W172, W173, I218).13 For instance, I218’s aliphatic side chain undergoes Van der Waals “packing” interactions with nearby CoQ alkyl groups 3.4 and 6.5 Å away. The fact that these nonpolar residues are located adjacent to the nonpolar face of the quinone ring is supported by the hydrophobic effect, a stabilizing force that drives hydrophobic alkyl groups to a protein’s interior by increasing the entropy of surrounding water molecules. This indicates that the Van der Waal forces described above are favorable, and that these hydrophobic interactions contribute to the stabilization of CoQ binding in SDH.13 Favorable binding of CoQ is significant to metabolism because it allows for the ETC to properly function, and in doing so, contributes to the production of ATP for cellular use.
Succinate dehydrogenase appears to exhibit distinct secondary structural motifs that contribute to cofactor binding. One such structure is the helix-loop-helix bundle, normally found in transcription factors, located between residues G446 and R542 in SDHA.14 This bundle is characterized by 4 α-helices, each connected by a loop, and is found at the succinate binding site. This helix-loop-helix motif may allow SDH to favorably interact and coordinate FAD at the active site. For example, hydrogen bonds from N527 (5.3 Å away) and E524 (6.5 Å away) with adenine’s purine amine group may allow for further stabilization of FAD in SDHA.6
Succinate Dehydrogenase Inhibition - A Structural & Environmental Analysis
Succinate dehydrogenase inhibitor (SDHI) fungicides play an important role in plant protection against many phytopathogenic fungi, and have recently been used as a cancer therapeutic strategy.15,16 Among the various toxic compounds that target diverse mitochondrial functions, many organisms, including humans, produce a subgroup of compounds that may target SDH.2 The mechanism of SDHIs is to inhibit the activity of SDH, thereby prohibiting mitochondrial respiration and ultimately killing pathogenic fungi.17 The need for SDHIs arose due to the increase in severity and scale of fungal disease in crops since the mid-20th century, as they initially presented a serious threat to food production and security.17 Specifically, fungal disease destroys essential calorie crops such as rice and maize, while also injuring commodity crops like coffee and bananas.17 Although various compounds were extensively used to reduce crop loss from these plant pathogens, many problems emerged with their use, including resistance and toxicity.17 Thus, over the course of the last 60 years, agro-industrial research programs have led to the development of structurally unique crop-protection compounds with many desirable properties including specificity, eradicant action and high activity at low use rates.15 For these reasons, SDHIs are now the second largest group of fungicides and have been widely applied to agricultural production, thereby becoming one of the fastest growing classes of fungicides since entering the market.17 After the development of 23 SDHIs as of 2020, it was determined that although all were diversified in structure, they all have similar structural skeletons.17 For example, all varieties share a carbonyl center, an amide, and an amine functional group.16 By characterizing these specific and potent inhibitors of SDH, researchers have been able to further their investigations of how the enzymes of aerobic metabolic processes function.18
There are three distinct classes of SDHIs: those that bind in the succinate pocket, the ubiquinone pocket, or by other means. Reversible inhibitors that competitively act at the succinate binding site include the citric acid cycle intermediates oxaloacetate and malate.16 These intermediates contribute to modulation of SDH activity according to cellular metabolic needs; that is, if cells already have produced significant amounts of energy via energy carriers like NADH, then these intermediates will subject SDHA to feedback inhibition and downregulate the citric acid cycle.16 Crystal structure shows that malate favorably binds via hydrogen bonds and charge-charge interactions with various polar SDHA residues.16 However, most existing SDHIs, including carboxin and atpenin A5, deactivate SDH by binding to the ubiquinone binding site at the SDHB, SDHC and SDHD subunits.17 Ubiquinone binding inhibitors specifically disrupt the reduction of ubiquinone to ubiquinol by blocking electron transport from the [3Fe-4S] cluster to ubiquinone.15 Carboxin was introduced in 1966 and was the first commercial fungicide targeting SDH in order to control seed growth, smut on wheat crops, and rust.17 More recently, other ubiquinone analogs that can inhibit a range of plant pathogens have been developed including boscalid, penthiopyrad and fluopyram.15 While all of these compounds reversibly affect activity, 3-nitropropionate is an irreversible, “suicide” inhibitor of SDH that displays antimycobacterial properties.2 This inhibitor is used in many ways, from skincare, to generating models of Huntington’s disease in rats.2 The location of the binding site of CoQ inhibitors was confirmed via X-ray crystallography by viewing their locations in chicken and pig mitochondrial SDH.13
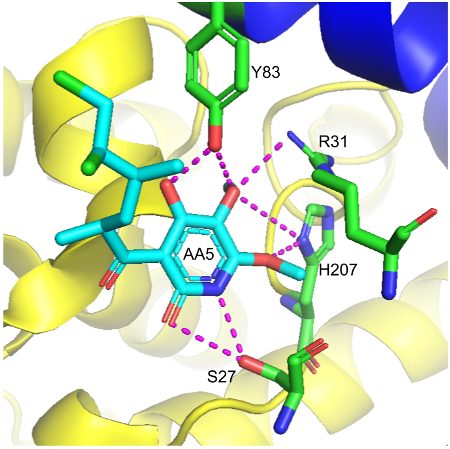
Although carboxin has been used extensively to understand the structure and function of SDH, high concentration is required for sufficient inhibition.18 This led to the discovery of atpenin A5 (AA5), an antifungal competitive inhibitor of SDH that is an analogue of ubiquinone.18 The use of atpenin A5 has been shown to produce an unpredictable cardioprotective effect, which is attributed to the potential activation of mitochondrial K+ channels.2 X-ray crystallography was used to confirm atpenin A5 inhibition in the E. coli SDH enzyme.13 As shown in Figure 3, binding of atpenin A5 occurs such that its polar face forms favorable hydrogen bonds with the side chains of polar hydrophilic residues of SDHB (H207), SDHC (S27, R31), and SDHD (Y83).3 Crystal structure has also shown that the long tail of AA5 extends out through the mouth of the binding site pocket, which presumably resembles CoQ with its ten isoprenoid units.13 This data encouraged biochemists to test for the strength of atpenin A5 SDH inhibition relative to other known inhibitors.18 Figure 4 shows that in bovine heart mitochondria, atpenin A5 is significantly more potent than the other SDHIs.18 For example, the IC50 value of atpenin A5 is 300-fold lower than that for carboxin (IC50 = 1.1 μM).18 Furthermore, atpenin A5 achieved complete (100%) inhibition of SDH at 0.1–1 μM, a concentration 2–3 orders of magnitude lower than that for carboxin.18 These findings reveal that both the structure of the aromatic ring and alkyl side chain are important for the potency of atpenin A5 to its binding site.
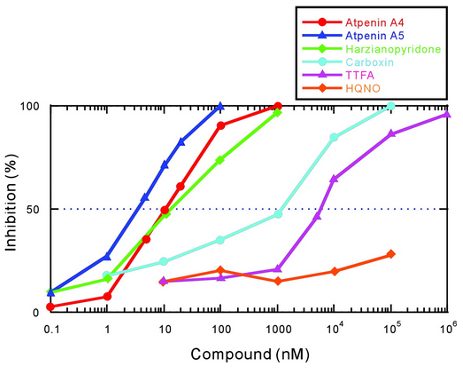
The discovery of atpenin A5’s high potency for binding SDH has encouraged the agrochemical industry to recommend the widespread use of SDHIs in order to counteract fungi proliferation on seeds and plants.2 As a result, in just a few decades, SDHIs are now omnipresent throughout the entire biosphere.2 This has caused major biodiversity loss and devastating ecological consequences as illustrated by the major collapse of insect and bird populations.2 The impregnation of SDHIs into living organisms may also play a role in the evolution of various human diseases, where mitochondrial, especially SDH, activity is impaired.2 For instance, thenoyltrifluoroacetone (TTFA) was shown to induce cell apoptosis in neuroblastoma cells.16 Although the human application of SDH inhibitors is still in its early stages (due to their potential for cytotoxicity), the generalized use of SDHIs is known to result in the rapid appearance of numerous mutated and resistant fungal species.2 In the future, these microorganisms could become multi-drug resistant, leading to the application of inhibitors with broader targets.2 This may worsen biodiversity loss through interference with other CoQ-binding proteins, such as those involved in complex III.2 This positive feedback loop of resistance to SDHIs makes the long-term use of SDHIs problematic.
The Role of Succinate Dehydrogenase Deficiency in Disease Development
Due to its fundamental role in the ETC and the citric acid cycle, succinate dehydrogenase is vital in most multicellular organisms. In order to properly function, SDH requires the proper folding and structure of its four subunits.1 This indicates that deleterious mutations in any subunit will invariably result in destabilization of the entire enzyme complex and decreased SDH activity.19 This has led to many investigations of the role of SDH deficiency on disease development. One study found that mutations causing mitochondrial SDH deficiency are not only lethal at the embryonic stage in mice, but are also observed post mortem in the brains of patients with Huntington’s Disease.20 These mutations have been identified in several types of cancer and have been shown to contribute to an abnormal accumulation of succinate in the cytosol of tumoral cells.19 SDH immunohistochemistry (IHC) is a useful diagnostic tool to triage patients for screening of any SDH mutation, where the stains of each subunit may indicate a correlation between SDH deficiency and disease development.19 Although the first mutation in an SDH gene (SDHD) was reported in patients with paraganglioma in 2000 via IHC, a new, in vivo approach has been developed to confirm the SDH-mutated status of a tumor by proton magnetic resonance spectroscopy.2 This method tests for succinate accumulation resulting from SDH inactivation.2 These tests indicated a correlation between SDH deficiency and paraganglioma, Leigh syndrome and clear cell renal cell carcinoma (ccRCC), conditions that can both be malignant or benign.5,16 This implies that in addition to its metabolic role, SDH also functions as a tumor suppressor.19
Germline mutations in the genes of SDHB, SDHC or SDHD can cause familial paraganglioma.5 Paragangliomas (PGLs) are rare neuroendocrine tumors that usually form in parasympathetic and sympathetic paraganglia, from the skull base to the pelvic region.2 Today, it is believed that 40% of PGLs are inherited, and that about half of these carry a germline mutation in the genes encoding SDH.2 PGL typically results from the increased production of superoxide ions, which leads to DNA damage and tumorigenesis.16 The two most common types of SDH deficient PGLs are pheochromocytoma and head and neck PGLs. Pheochromocytoma tumors are more aggressive with increased metastasis, mortality, and secretion of catecholamines from the sympathetic chain.2 On the other hand, when they occur as head and neck PGLs from the parasympathetic chain, these tumors are benign and rarely secrete catecholamines.5 Both types of tumors can occur as young as 10 years old.5 While the incidence of PGLs in the healthy public is relatively low, 70% of individuals with underlying SDHB, SDHC, or SDHD mutations will develop PGLs by 80 years old.5 PGL malignancy varies based on which SDH subunit in the patient mutates. For example, PGLs due to SDHB mutants are commonly malignant and are localized to the adrenal glands, while tumors caused by SDHC and SDHD mutants are almost always benign and can be found in the head and neck.19
While mutations in SDHB, SDHC and SDHD are commonly attributed to PGLs, SDHA mutations are most likely to lead to Leigh syndrome, an early-onset progressive neurodegenerative disorder.5 Patients with Leigh syndrome typically experience symptoms in early infancy such as developmental delay, weakness, seizures and vomiting.5 These symptoms are due to a sporadic or inherited metabolic dysfunction of the mitochondria.5 There is no known cure for Leigh syndrome, and patients often die from their disease within several months of being diagnosed.5 One study found that in two siblings with Leigh syndrome, both were homozygous for an R554W substitution in the SDHA subunit, causing SDH deficiency.21 The deleterious effect of the Arg to Trp substitution on the catalytic activity of SDH was also observed in a SDH deficient yeast strain transformed with mutant SDHA cDNA.21 This case report was important because it was the first time in humans that a nuclear gene mutation (SDHA) was found to cause a mitochondrial respiratory chain deficiency.5 Although these genetic changes disrupt the activity of SDH, it is unknown how mutations in the SDHA gene are related to the specific features of Leigh syndrome.21
Clear cell renal cell carcinoma (ccRCC) is the most common type of kidney cancer, accounting for ∼80% of all kidney cancers.4 Despite recent advances, ccRCC is incurable and highly malignant, with a 5 year survival rate of <20%. After recognizing that further biologic and therapeutic insights in this disease were needed, researchers found that reduced SDH activity, resulting in adverse succinate accumulation, is a common characteristic in ccRCC with a direct impact on survival in patients.4 Figure 5A-C shows that SDH subunits SDHB, SDHC and SDHD are significantly downregulated in ccRCC tumors (n = 573) relative to normal renal tissue (n = 72). Moreover, survival analyses from the patient sample revealed a markedly worse overall survival as a direct consequence of lower SDHB, SDHC and SDHD expression (Fig. 5). This supports a direct trend between SDH expression and overall patient survival in ccRCC patients.4
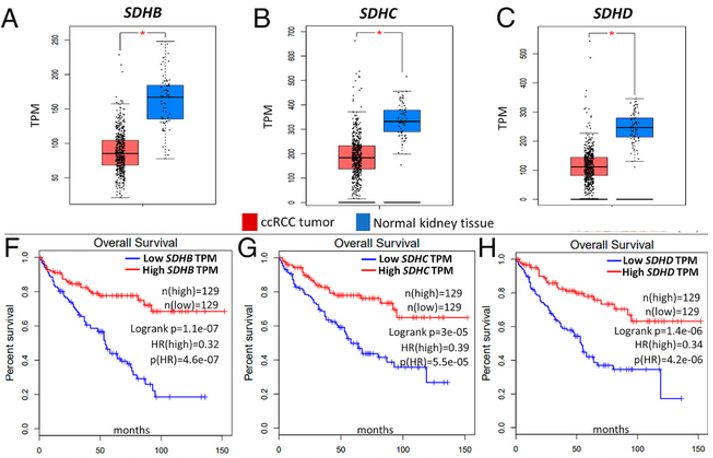
While the precise pathway to tumorigenesis is not yet fully elucidated, there are various theories regarding the cause of SDH deficiency.19 According to the Knudson two-hit hypothesis, as a tumor suppressor gene, both alleles of SDH must be inactivated in order to cause a loss of heterozygosity and thereby halt the production of SDH protein.5,22 Moreover, it is suspected that malfunction of the SDH complex due to mutations from the SDHB, SDHC, and SDHD genes can cause a hypoxic response in the cell that leads to tumor formation.19 Because the mutated SDH enzyme cannot convert succinate to fumarate, succinate accumulates in the cytosol.19 Excess succinate then leads to increased reactive oxygen species (ROS) production, which abnormally triggers the hypoxia pathways and stabilizes hypoxia-inducible factors (HIF).19 This stimulates cells to divide rapidly and uncontrollably, which can lead to the development of tumors in people with hereditary PGL.19 Other studies have hypothesized that SDH mutation causes tumorigenesis through increased histone methylation via inhibition of histone demethylase JumonjiD316, impaired developmental apoptosis via inhibition of prolyl hydroxylase Eg1N323, and a nonsense mutation in the mRNA transcript for SDHB at nucleotide position 136.24 This results in translation of a truncated SDHB protein, which is further enhanced by hypoxia.24
Conclusion
Over the past 50 years, the SDH complex has been the subject of renewed interest by biochemists because of its critical role in aerobic metabolism in eukaryotes. SDH is a heterotetrameric, transmembrane protein complex bound to the inner mitochondrial membrane that links the citric acid cycle to the ETC. Crystal structure of SDH, discovered via X-ray crystallography, shows that the SDHA and SDHB subunits form the catalytic domain and bind succinate, while the SDHC and SDHD subunits are integral proteins that bind ubiquinone (CoQ). Various bound cofactors and prosthetic groups, including FAD, iron-sulfur centers and hemes stabilize SDH assembly and enhance catalytic electron transport. While inhibitors of SDH (SDHIs) were first developed to combat fungal disease within crops in the mid-20th century, their modern widespread use is directly correlated with severe ecological harm, as shown by the major collapse of insect and bird populations. Most SDHIs reversibly and competitively bind in the succinate pocket (oxaloacetate, malate) or in the ubiquinone pocket (carboxin, atpenin A5) with varying efficiency. Both ligand and inhibitor binding is supported through hydrogen bonds, Van der Waals “packing” forces and the hydrophobic effect with nearby residues in SDH. Lastly, because of advancements in screening techniques like immunohistochemistry, deleterious mutations that cause SDH deficiency have been shown to play a role in the evolution of various human diseases, including paraganglioma, Leigh syndrome and ccRCC.
Although this review reveals significant insight into SDH’s function and clinical application from the works of many scientists, these discoveries supply more questions than answers. For instance, investigations on how different SDH mutants influence ROS production may provide new clues that can explain the role of mitochondrial ROS in cellular tumorigenesis and mutant phenotypes. Moreover, the effects of SDH inhibition on the growth of human cells have yet to be fully understood. While early trials suggest that SDHIs can function toward cancer therapy, it is possible that in populations with increased susceptibility to oxidative stress, SDHIs could accelerate disease progression. Thus, more research must be done to assess the toxicity of SDHIs in humans. Lastly, the widespread use of SDHIs in the agrochemical industry has caused the rapid appearance of resistant fungal species. The fact that this has further advanced fungi proliferation in crops is a challenge for the development of new SDHIs. Future implementation of a dual-target drug design and computer-aided synthesis strategies may therefore help to develop safer SDHI fungicides that delay drug resistance and prevent fungal diseases.
References
(1) Huang, S.; Millar, A. H. Succinate Dehydrogenase: The Complex Roles of a Simple Enzyme. Curr. Opin. Plant Biol. 2013, 16 (3), 344–349. https://doi.org/10.1016/j.pbi.2013.02.007.
(2) Bénit, P.; Goncalves, J.; El Khoury, R.; Rak, M.; Favier, J.; Gimenez-Roqueplo, A.-P.; Rustin, P. Succinate Dehydrogenase, Succinate, and Superoxides: A Genetic, Epigenetic, Metabolic, Environmental Explosive Crossroad. Biomedicines 2022, 10 (8), 1788. https://doi.org/10.3390/biomedicines10081788.
(3) Horsefield, R.; Yankovskaya, V.; Sexton, G.; Whittingham, W.; Shiomi, K.; Ōmura, S.; Byrne, B.; Cecchini, G.; Iwata, S. Structural and Computational Analysis of the Quinone-Binding Site of Complex II (Succinate-Ubiquinone Oxidoreductase): A MECHANISM OF ELECTRON TRANSFER AND PROTON CONDUCTION DURING UBIQUINONE REDUCTION*. J. Biol. Chem. 2006, 281 (11), 7309–7316. https://doi.org/10.1074/jbc.M508173200.
(4) Aggarwal, R. K.; Luchtel, R. A.; Machha, V.; Tischer, A.; Zou, Y.; Pradhan, K.; Ashai, N.; Ramachandra, N.; Albanese, J. M.; Yang, J.; Wang, X.; Aluri, S.; Gordon, S.; Aboumohamed, A.; Gartrell, B. A.; Hafizi, S.; Pullman, J.; Shenoy, N. Functional Succinate Dehydrogenase Deficiency Is a Common Adverse Feature of Clear Cell Renal Cancer. Proc. Natl. Acad. Sci. 2021, 118 (39), e2106947118. https://doi.org/10.1073/pnas.2106947118.
(5) Rutter, J.; Winge, D. R.; Schiffman, J. D. Succinate Dehydrogenase – Assembly, Regulation and Role in Human Disease. Mitochondrion 2010, 10 (4), 393–401. https://doi.org/10.1016/j.mito.2010.03.001.
(6) Huang, L.; Sun, G.; Cobessi, D.; Wang, A. C.; Shen, J. T.; Tung, E. Y.; Anderson, V. E.; Berry, E. A. 3-Nitropropionic Acid Is a Suicide Inhibitor of Mitochondrial Respiration That, upon Oxidation by Complex II, Forms a Covalent Adduct with a Catalytic Base Arginine in the Active Site of the Enzyme *. J. Biol. Chem. 2006, 281 (9), 5965–5972. https://doi.org/10.1074/jbc.M511270200.
(7) Kenney, W. C. The Reaction of N-Ethylmaleimide at the Active Site of Succinate Dehydrogenase. J. Biol. Chem. 1975, 250 (8), 3089–3094.
(8) Robinson, K. M.; Lemire, B. D. Covalent Attachment of FAD to the Yeast Succinate Dehydrogenase Flavoprotein Requires Import into Mitochondria, Presequence Removal, and Folding (∗). J. Biol. Chem. 1996, 271 (8), 4055–4060. https://doi.org/10.1074/jbc.271.8.4055.
(9) Lancaster, C. R.; Kröger, A. Succinate: Quinone Oxidoreductases: New Insights from X-Ray Crystal Structures. Biochim. Biophys. Acta 2000, 1459 (2–3), 422–431. https://doi.org/10.1016/s0005-2728(00)00180-8.
(10) Kim, H. J.; Khalimonchuk, O.; Smith, P. M.; Winge, D. R. Structure, Function, and Assembly of Heme Centers in Mitochondrial Respiratory Complexes. Biochim. Biophys. Acta 2012, 1823 (9), 1604–1616. https://doi.org/10.1016/j.bbamcr.2012.04.008.
(11) Yankovskaya, V.; Horsefield, R.; Törnroth, S.; Luna-Chavez, C.; Miyoshi, H.; Léger, C.; Byrne, B.; Cecchini, G.; Iwata, S. Architecture of Succinate Dehydrogenase and Reactive Oxygen Species Generation. Science 2003, 299 (5607), 700–704. https://doi.org/10.1126/science.1079605.
(12) Brandt, U.; Trumpower, B. The Protonmotive Q Cycle in Mitochondria and Bacteria. Crit. Rev. Biochem. Mol. Biol. 1994, 29 (3), 165–197. https://doi.org/10.3109/10409239409086800.
(13) Huang, L.; Lümmen, P.; Berry, E. A. Crystallographic Investigation of the Ubiquinone Binding Site of Respiratory Complex II and Its Inhibitors. Biochim. Biophys. Acta BBA - Proteins Proteomics 2021, 1869 (9), 140679. https://doi.org/10.1016/j.bbapap.2021.140679.
(14) Jones, S. An Overview of the Basic Helix-Loop-Helix Proteins. Genome Biol. 2004, 5 (6), 226. https://doi.org/10.1186/gb-2004-5-6-226.
(15) Avenot, H. F.; Michailides, T. J. Progress in Understanding Molecular Mechanisms and Evolution of Resistance to Succinate Dehydrogenase Inhibiting (SDHI) Fungicides in Phytopathogenic Fungi. Crop Prot. 2010, 29 (7), 643–651. https://doi.org/10.1016/j.cropro.2010.02.019.
(16) Moreno, C.; Santos, R. M.; Burns, R.; Zhang, W. C. Succinate Dehydrogenase and Ribonucleic Acid Networks in Cancer and Other Diseases. Cancers 2020, 12 (11), 3237. https://doi.org/10.3390/cancers12113237.
(17) Li, S.; Li, X.; Zhang, H.; Wang, Z.; Xu, H. The Research Progress in and Perspective of Potential Fungicides: Succinate Dehydrogenase Inhibitors. Bioorg. Med. Chem. 2021, 50, 116476. https://doi.org/10.1016/j.bmc.2021.116476.
(18) Miyadera, H.; Shiomi, K.; Ui, H.; Yamaguchi, Y.; Masuma, R.; Tomoda, H.; Miyoshi, H.; Osanai, A.; Kita, K.; Ōmura, S. Atpenins, Potent and Specific Inhibitors of Mitochondrial Complex II (Succinate-Ubiquinone Oxidoreductase). Proc. Natl. Acad. Sci. 2003, 100 (2), 473–477. https://doi.org/10.1073/pnas.0237315100.
(19) Dalla Pozza, E.; Dando, I.; Pacchiana, R.; Liboi, E.; Scupoli, M. T.; Donadelli, M.; Palmieri, M. Regulation of Succinate Dehydrogenase and Role of Succinate in Cancer. Semin. Cell Dev. Biol. 2020, 98, 4–14. https://doi.org/10.1016/j.semcdb.2019.04.013.
(20) Skillings, E. A.; Morton, A. J. Delayed Onset and Reduced Cognitive Deficits through Pre-Conditioning with 3-Nitropropionic Acid Is Dependent on Sex and CAG Repeat Length in the R6/2 Mouse Model of Huntington’s Disease. 2016. https://doi.org/10/254599.
(21) Bourgeron, T.; Rustin, P.; Chretien, D.; Birch-Machin, M.; Bourgeois, M.; Viegas-Péquignot, E.; Munnich, A.; Rötig, A. Mutation of a Nuclear Succinate Dehydrogenase Gene Results in Mitochondrial Respiratory Chain Deficiency. Nat. Genet. 1995, 11 (2), 144–149. https://doi.org/10.1038/ng1095-144.
(22) Xekouki, P.; Pacak, K.; Almeida, M.; Wassif, C. A.; Rustin, P.; Nesterova, M.; de la Luz Sierra, M.; Matro, J.; Ball, E.; Azevedo, M.; Horvath, A.; Lyssikatos, C.; Quezado, M.; Patronas, N.; Ferrando, B.; Pasini, B.; Lytras, A.; Tolis, G.; Stratakis, C. A. Succinate Dehydrogenase (SDH) D Subunit (SDHD) Inactivation in a Growth-Hormone-Producing Pituitary Tumor: A New Association for SDH? J. Clin. Endocrinol. Metab. 2012, 97 (3), E357–E366. https://doi.org/10.1210/jc.2011-1179.
(23) Lee, S.; Nakamura, E.; Yang, H.; Wei, W.; Linggi, M. S.; Sajan, M. P.; Farese, R. V.; Freeman, R. S.; Carter, B. D.; Kaelin, W. G.; Schlisio, S. Neuronal Apoptosis Linked to EglN3 Prolyl Hydroxylase and Familial Pheochromocytoma Genes: Developmental Culling and Cancer. Cancer Cell 2005, 8 (2), 155–167. https://doi.org/10.1016/j.ccr.2005.06.015.
(24) Baysal, B. E. A Recurrent Stop-Codon Mutation in Succinate Dehydrogenase Subunit B Gene in Normal Peripheral Blood and Childhood T-Cell Acute Leukemia. PLoS ONE 2007, 2 (5), e436. https://doi.org/10.1371/journal.pone.0000436.