In a rare disease called mucolipidosis type II, people’s hearts and abdomens swell, and their bones grow malformed.
A lysosomal storage disorder, mucolipidosis type II causes edema of the internal organs and skeletal dysplasia. Children diagnosed with the genetic disease often die before they reach age 7. Now, University of Michigan researchers have identified a new gene implicated in the disease, TMEM251, which is necessary for lysosomes to function correctly.
Lysosomes are organelles within all cells of the body—except red blood cells—responsible for taking in and recycling the garbage your cells produce. When the lysosome can’t function properly, it fails to recycle this garbage and instead simply stockpiles them in the organelle.

The team, led by Ming Li, assistant professor of molecular, cellular and developmental biology, discovered that if TMEM251 is defective, it fails to encode the pathway for the enzymes necessary for the correct function of lysosomes to travel inside the lysosome. The study is published in Nature Communications.
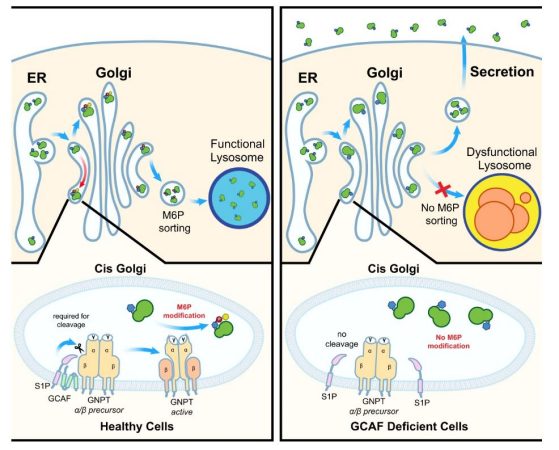
There are about 50 to 60 enzymes inside the lysosomes that digest worn-out cellular parts as well as waste from outside the cell. The lysosome also recycles this waste—proteins, nucleic acid, carbohydrates and lipids—back into usable material. But for these enzymes to travel inside the lysosome, they need a signal called the mannose-6-phosphate biosynthetic pathway, or M6P.
“It’s like a postage stamp. The enzymes have to have this signal in order to go inside the lysosome. If they don’t have M6P, they aren’t able to go into the lysosome,” Li said. “So consequently, you still have lysosomes, but not a single one of them would be functional because they lack these enzymes.”
Li’s lab studies the lysosome, and in particular, the composition of lysosome membrane proteins. The lysosome has the ability to regulate its own membrane protein by triggering the degradation of these proteins through a process called ubiquitination. This process allows proteins to travel from the membrane of the lysosome inside the organelle for degradation. The researchers also wanted to understand which genes were responsible for lysosome function and what happens when those genes are defective.
To do this, the team used a CRISPR knock-out screen that knocked out every gene in the human genome at the cellular level, one by one. The researchers were then able to study what happens in the lysosome in response to each gene’s deletion. Specifically, the researchers were looking for genes that could be responsible for the lysosome’s degradation.
The experiment turned up TMEM251.
“So then the game became, why is this gene so important for human health? And why is it so critical for lysosomal function?” Li said.
The group discovered that the TMEM251 gene encodes an enzyme that activates M6P, a pathway that most of the 50 to 60 digesting enzymes in the lysosomes require. In a literature review, the researchers also found a 2021 paper that described mucolipidosis type II-like symptoms in humans that result from having a defective TMEM251 gene.
“Our discovery answered the molecular mechanism of this new human disease,” Li said.
The protein encoded by the gene TMEM251 is required to activate another enzyme called GNPT, which catalyzes the M6P pathway. The researchers also demonstrated that TMEM251 is localized to the Golgi apparatus, a structure that forms lysosomes. That the two enzymes are localized to the Golgi fits the idea that the proteins have to work together to add M6P to lysosomal enzymes, Li said. The researchers named TMEM251 as GNPT cleavage and activity factor, or GCAF.
The researchers then checked what would happen if they knocked out the TMEM251 gene in zebrafish. Comparing the wild-type zebrafish with the zebrafish whose TMEM251 gene had been knocked out, the researchers could see defects to the zebrafish’s abdomen, skeletal and cartilage development and heart.
Co-author Xi Yang said the team also suggests a therapy strategy to combat the illness in humans. The therapy, which is in very early stages, is based on what they call an “enzyme replacement therapy.” The researchers demonstrated that if they supplied the enzyme which contains the M6P modification to TMEM251-deficient cells, that enzyme was able to filter into the cell through a process called endocytosis and be delivered to a malfunctioning lysosome.
“We know that the pathogenesis of this disease is because you don’t have a functional lysosome,” said Yang, a research specialist in Li’s lab. “This knockout cell can actually use these endocytosed functional enzymes to rebuild their lysosome and make it functional again. You can rescue the deficiency, at least at a cellular level.”
The team recently received a National Institutes of Health grant to further study the TMEM251 gene, particularly how the TMEM251 enzyme interacts with the GNPT enzyme to facilitate the genesis of M6P. The team also aims to describe what TMEM251 looks like at the structural level.
Co-authors of the paper include U-M MCDB’s Weichao Zhang, Linchen Yu, Bokai Zhang, Jianchao Zhang, Varsha Venkatarangan, Liang Chen, Sarah Bui and Yanzhuang Wang. MCDB professor Cunming Duan and postdoctoral researcher Yingxiang Li assisted in the zebrafish work. Woo Yung Cho joined the team from the BRCF Microscopy Core at the U-M Medical School. Bala Bharathi Burugula and Jacob Kitzman with the U-M Medical School Department of Human Genetics also contributed.
-- written by Morgan Sherburne, University of Michigan News